
This blog is adapted from
ProCyon: A multimodal foundation model for protein phenotypes
ProCyon: A Multimodal Foundation Model for Protein Phenotypes
December 19, 2024Understanding protein phenotypes is a fundamental challenge in biology, crucial for decoding how living systems operate at the molecular level and beyond. Artificial intelligence models have enabled leaps in biology, with breakthroughs like protein structure prediction (AlphaFold), protein sequence modeling (ESM), and variant effect prediction (AlphaMissense). Predicting protein structure from sequence is now significantly advanced, however, predicting protein phenotypes—the observable characteristics that connect molecular functions to biological roles—using sequence and/or structure remains an open challenge.
Approximately 20% of human proteins remain entirely uncharacterized, and even well-annotated proteins often lack functional insights across biological contexts and disease states, and 40% of human proteins are missing context-specific functional insights. Research bias further compounds this challenge: 95% of life science publications focus on only 5,000 proteins, leaving vast portions of the human proteome uncharted.
We developed ProCyon, a groundbreaking 11-billion-parameter unified model of protein phenotypes designed to model, generate, and predict phenotypes across five interrelated knowledge domains: molecular functions, therapeutic mechanisms, disease associations, functional protein domains, and molecular interactions.
ProCyon bridges multimodal protein representations and large language models (LLMs). Modeling protein phenotypes requires a multimodal approach, meaning that natural language descriptions of phenotypes, protein sequence, or protein structure alone are insufficient to fully represent phenotypes. Text-based LLMs face challenges annotating proteins with phenotypes due to biases in the scientific literature, which tends to emphasize well-studied proteins. These models also lack explicit representations of proteins. For example, LLMs understand proteins through standardized naming systems, such as the HUGO Gene Nomenclature Committee. However, poorly annotated proteins often lack sufficient coverage in the literature, limiting their representation in LLM training corpora. This gap can be addressed by protein representation learning encoders that operate directly on protein sequences and/or structures to determine a protein’s function. Protein encoders, such as protein language models for sequences and geometric deep learning methods for structures, excel at generating generalizable protein representations. However, these methods are constrained by reliance on pre-defined functional categories, which limits their scalability to phenotypes of arbitrary complexity. Addressing this challenge requires a general solution that integrates the strengths of LLMs and protein encoders to enable new insights into the human proteome.

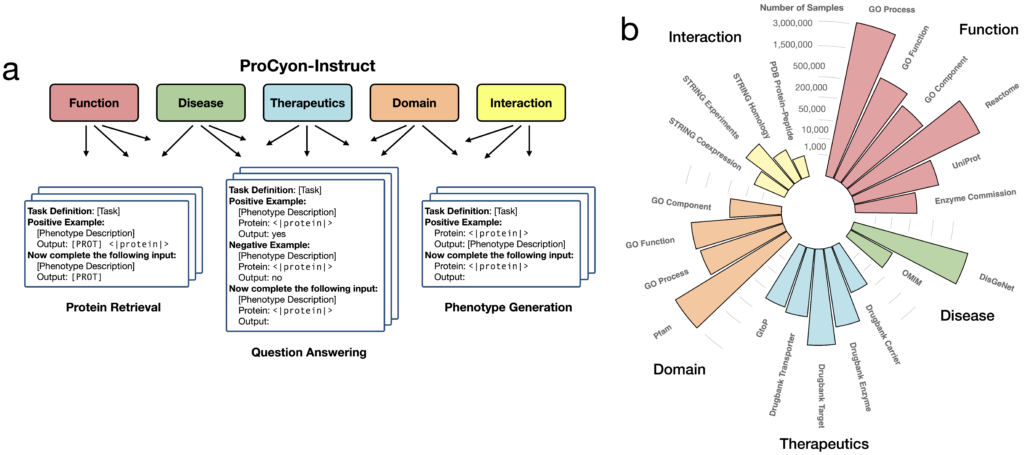
Overview of ProCyon model architecture
We train ProCyon to follow human instructions tailored to protein annotation, employing three core approaches. In Protein retrieval (Figure 1a), ProCyon converts a phenotype description into a ranked list of proteins matching the user’s prompt. For Question answering (Figure 1b), ProCyon processes interleaved multimodal inputs, including protein and phenotype data, to determine whether the protein aligns with the phenotype. In Phenotype generation (Figure 1c), ProCyon takes a prompt about a specific protein and produces a free-text description of the protein’s phenotype, conditioned on a user-defined biological context.
ProCyon’s architecture and inference mechanisms are designed to tackle the challenges of protein phenotypes. The model integrates interleaved inputs of protein sequences, structures, and natural language using a multimodal token composition algorithm. These inputs are processed through combined protein sequence and structure encoders, which feed rich multimodal representations into a pretrained LLM. ProCyon also addresses the one-to-many problem of protein-phenotype prediction: a single protein may participate in multiple distinct biological processes, and a single phenotype often involves numerous related proteins.
To tackle this complexity, ProCyon uses a multifaceted approach. Training across multiple knowledge domains achieves multiscale phenotypic resolution, enabling precise querying of molecular functions, diseases, pathways, and therapeutics. Additionally, a novel context augmentation strategy enriches instruction prompts with supplemental phenotype information, improving predictive accuracy. ProCyon also supports compositional queries, allowing users to dynamically define phenotypes by combining multiple traits into a single query—an approach far more flexible than static vocabularies used in traditional models. Finally, ProCyon’s inference mechanisms manage one-to-many relationships through contextual protein retrieval and conditional phenotype generation. These mechanisms include multi-protein prioritization and beam search sampling, ensuring robust and accurate phenotype predictions.
Zero-shot task transfer and dynamic task specification in ProCyon
In molecular biology, the complexity and uniqueness of most problems make it essential to move beyond predefined task sets. Traditional protein annotation methods rely on fixed, static tasks. For instance, Gene Ontology annotation predictors typically involve a model trained on protein sequences or structures, with classification heads producing predefined outputs. However, this rigid approach is not scalable for the broader scope of protein annotation, as it cannot account for the vast and evolving range of biological queries.
Different scientists often approach problems differently, requiring a model capable of accommodating diverse user expertise and adapting to prompts that are complex and multi-faceted, and it is impossible to anticipate the full space of queries at train time. Inspired by how LLMs process varied user inputs, we designed ProCyon to enable dynamic task specification—a paradigm shift in protein annotation. This capability allows scientists to define tasks dynamically using natural language prompts, enabling the model to generalize to entirely new tasks at inference time and tailor predictions to their specific needs.
Dynamic task specification refers to the model’s ability to switch tasks at inference by leveraging a universal representation of tasks through natural language prompts. This requires redefining task transfer—not simply as transferring across input instances or distributions but as adapting to entirely new tasks with unique conditional specifications. While the concept of task transfer is not new (see classic definitions of instruction tuning in Wei et al., 2022 or Wang et al., 2022), its application in protein annotation addresses the limitations of structured, constrained databases. Using instruction tuning to train ProCyon, we created a novel dataset, ProCyon-Instruct, designed to enable dynamic task specification.
ProCyon-Instruct dataset with 33 million protein-phenotype instructions
To power ProCyon, we built ProCyon-Instruct, a dataset that bridges five key knowledge domains: molecular functions, disease phenotypes, therapeutics, protein domains, and protein-protein interactions. By unifying diverse protein-phenotype pairs under a structured instruction tuning framework (Figure 2a), ProCyon-Instruct transforms raw data into interleaved natural language instructions that emulate how scientists approach protein analysis.
The innovation lies in addressing a fundamental limitation of existing databases: their reliance on templated, standardized language that lacks diversity and fails to generalize to real-world queries from users with varying expertise. Using task definitions, example descriptions, and phenotype descriptions as input prompts, we rephrase protein-phenotype descriptions along two critical dimensions:
- Rephrasing style: Direct rephrasing, summarization, or simplification.
- Expertise level: Tailored to junior scientists, experienced researchers, and expert scientists.
This approach dramatically expanded the linguistic richness of the dataset, producing 33 million protein-phenotype instructions. By leveraging this rephrasing system, ProCyon-Instruct enables ProCyon to generalize to out-of-distribution inputs and support nuanced natural language queries across varying contexts and expertise levels.
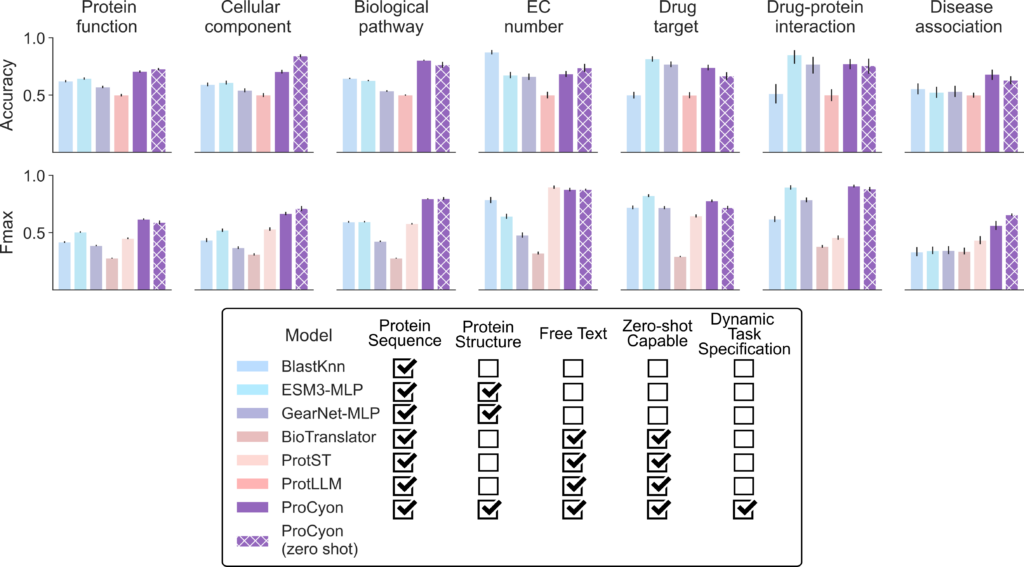
Benchmarking ProCyon against single-modality and multi-modality models
ProCyon consistently demonstrates exceptional performance across 14 types of tasks designed to evaluate contextual protein retrieval, question answering, and zero-shot generalization. When compared to single-modality models, ProCyon outperforms in 10 of 14 tasks. It achieves a question-answering accuracy of 72.7%, surpassing the best single-modality model, ESM3-MLP, which reaches 67.8% (Figure 3). ProCyon achieves a Fmax of 0.743 on protein retrieval tasks, well above the runner-up (ESM3-MLP, Fmax of 0.618).
ProCyon also excels against multimodal models, outperforming them in 13 of 14 tasks. For example, it achieves a 30.1% improvement in Fmax over ProtST, the next-best multimodal model, in protein retrieval tasks. In zero-shot retrieval, ProCyon achieves an average Fmax improvement of 27%, highlighting its ability to generalize effectively to unseen phenotypes. This robust performance across multiple knowledge domains underscores ProCyon’s unique ability to harmonize multimodal representations of sequence, structure, and text.
The model’s adaptability is further evident in its ability to process complex queries. For example, ProCyon effectively handles compositional prompts describing pleiotropic phenotypes, outperforming manual aggregation approaches by 3.97% in pathway crosstalk tasks.
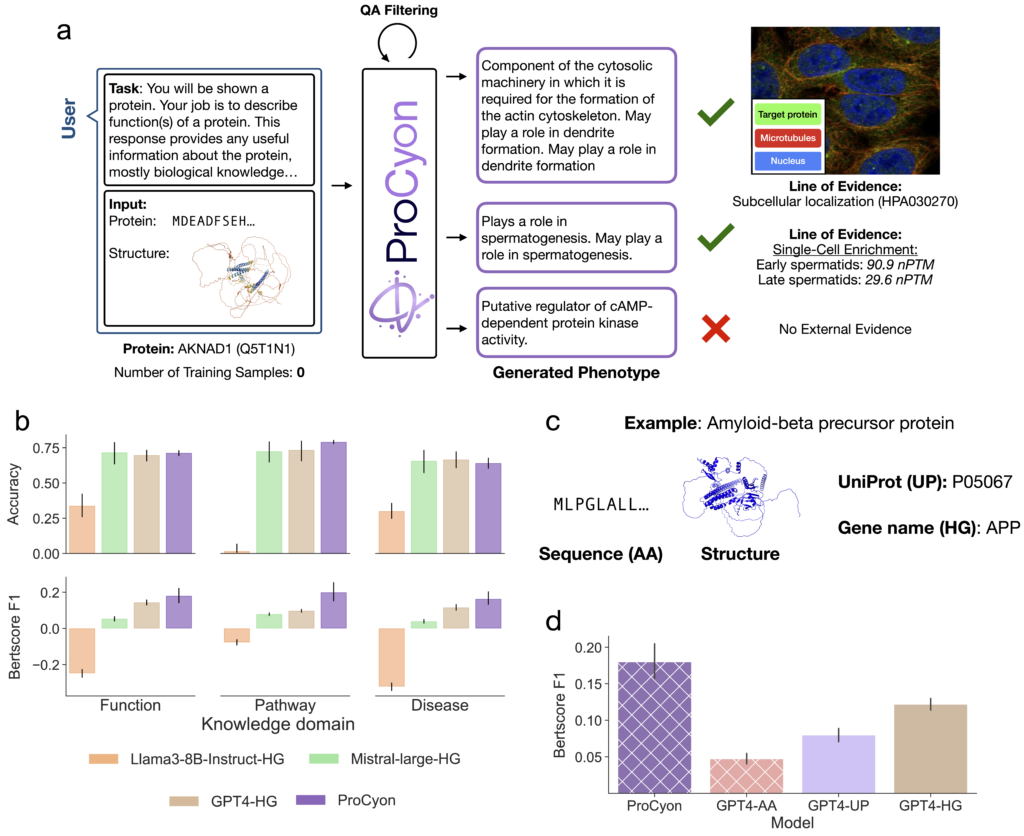
Generation of protein phenotypes
ProCyon excels at generating free-text descriptions of protein phenotypes, providing insights that are not constrained by pre-defined vocabularies or ontologies. This capability allows ProCyon to describe protein functions across molecular, cellular, and systemic scales. For instance, it predicts novel functions for poorly characterized proteins such as AKNAD1 (Figure 4a). This highlights ProCyon’s potential to illuminate the dark proteome—regions of human proteome with yet-undetermined functions.
To evaluate its phenotype generation abilities, ProCyon was benchmarked against text-only LLMs in protein-phenotype-centric tasks. ProCyon outperformed LLMs, even frontier models with far more parameters, in question answering and semantic similarity tasks (measured via BertScore), achieving higher alignment between generated text and reference texts across multiple knowledge domains (Figure 4b). These results demonstrate ProCyon’s unique ability to integrate multimodal data for precise and context-aware phenotype generation.
When comparing against text-only LLMs, we find that the method of encoding a protein in text is critical to their performance. For example, a protein could be encoded using its ID in different biological databases or by its raw protein sequence (Figure 4c). We experiment with two canonical protein IDs-–HUGO Gene Nomenclature Committee (HGNC or HG) and UniProt (UP)—and an encoding of proteins using their amino acid sequence (AA). A key point is that the full amino acid sequence is the only text encoding method that could generalize to arbitrary, possibly novel, proteins. GPT-4 performance is worst for AA and best for HG, but both methods perform worse than ProCyon for the BertScore metric (Figure 4d).
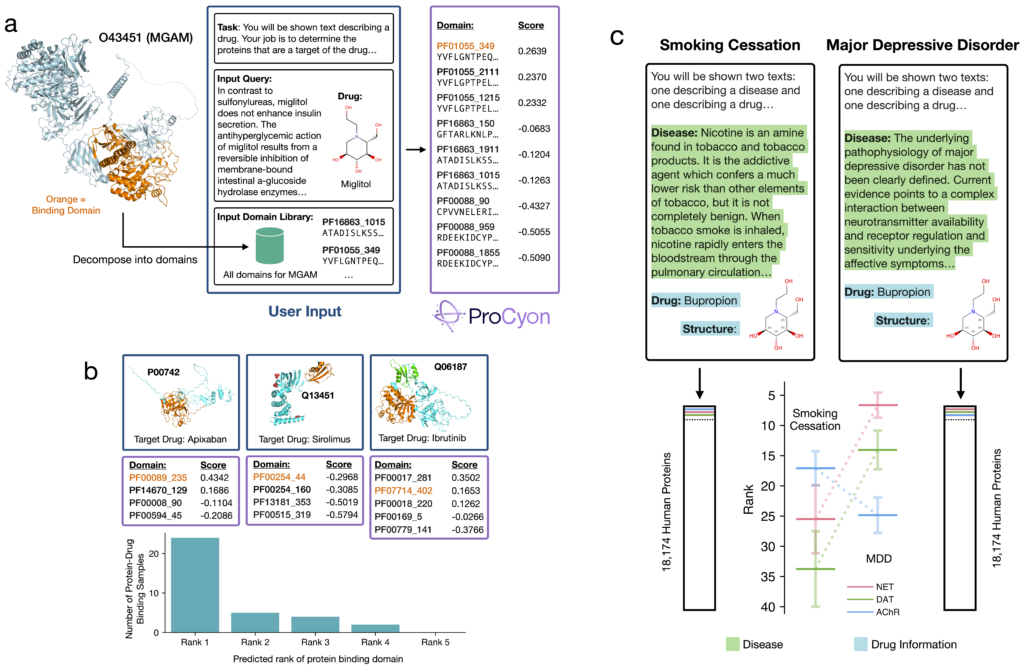
New applications in biology
We extensively evaluate ProCyon for biological applications, particularly through its ability to perform zero-shot tasks—solving tasks that were not explicitly present in its training data. One such application is the identification of protein domains that bind to small molecule drugs (Figure 5a). Despite not being trained on domain-level drug binding data, ProCyon accurately identifies the correct binding domain in 24 out of 35 cases (Figure 5b). This performance highlights ProCyon’s adaptability in tackling the complex ligand-binding prediction problem.
ProCyon also excels in retrieval tasks based on complex compositional queries across biological knowledge domains. For example, it differentiates between bupropion’s therapeutic mechanisms in treating major depressive disorder (MDD) and nicotine addiction (Figure 5c). When prompted for MDD, ProCyon retrieves the norepinephrine transporter (NET) as the primary target, while for nicotine addiction, it prioritizes the cholinergic receptor (AChR). This nuanced understanding demonstrates ProCyon’s ability to contextualize its predictions based on multi-scale biological inputs.
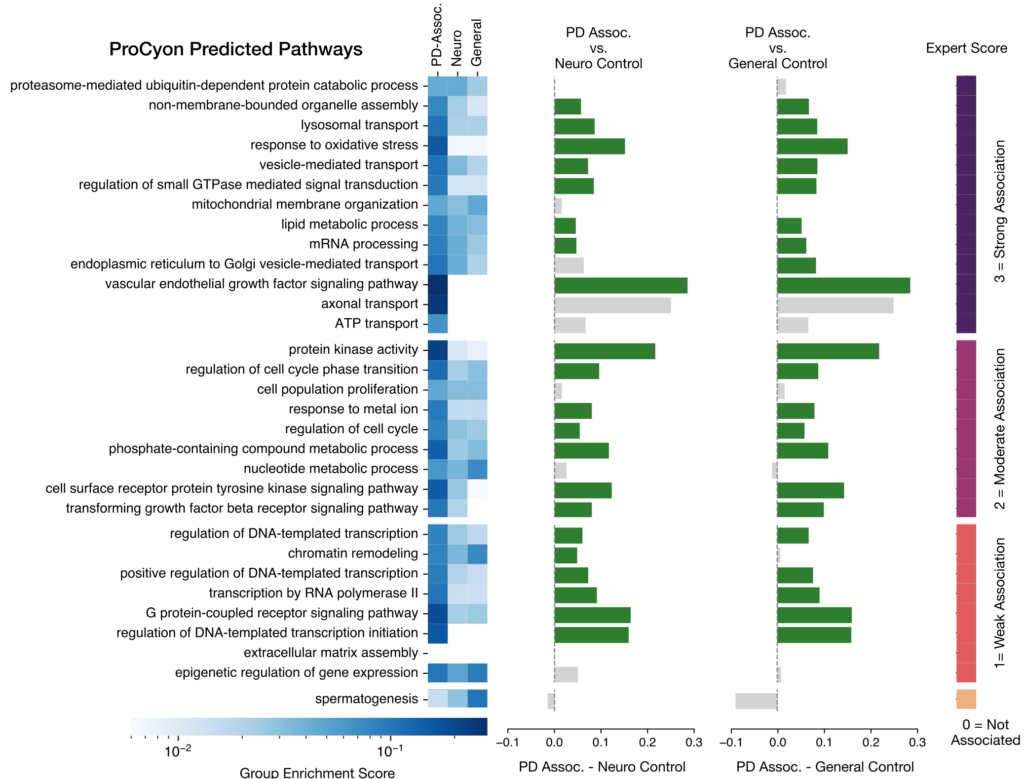
ProCyon can generate candidate phenotypes for poorly characterized proteins recently linked to Parkinson’s disease (PD). By producing detailed phenotype descriptions for these PD-associated proteins, ProCyon identified biological pathways that received high scores in expert reviews and showed significant enrichment in functional disease pathway analyses (Figure 6).
ProCyon also excels in protein-peptide binding, accurately distinguishing binders from non-binders for ACE2 using mass spectrometry datasets from peptide candidates, paving the way for advancements in peptide-based therapies. Additionally, ProCyon tackles complex phenotypes like pleiotropic roles and pathway crosstalk, including identifying STING’s role in neuronal inflammatory stress—an insight only recently characterized. Beyond these capabilities, ProCyon models the functional impacts of genetic mutations, effectively distinguishing between benign and pathogenic variants, such as PSEN1 mutations associated with Alzheimer’s disease, enabling deeper exploration of disease biology.
Conclusion
ProCyon represents a transformative step toward a universal solution for functional protein biology, offering the potential to unlock new insights into the human proteome. Its ability to perform zero-shot generalization, combined with an intuitive natural language interface, democratizes access to cutting-edge protein analysis tools. ProCyon is an open model, and we make our training datasets, training and inference code, and pretrained models publicly available to encourage future work and exploration.